This lecture can be viewed on YouTube site: Life Scinence Lectures for you
https://youtu.be/ocg37FwHteg
1. Introduction
This lecture explains the transcriptional repression of genes through methylation of cytosine within CpG sequences, as well as the demethylation reaction.
- Characteristics of 5mCpG methylation in animal cell genomes
- Frequency of C methylation in the human genome
- CpG suppression in genomes
- Transcriptional repression mechanism by methylated cytosine in eukaryotes
- Types and roles of DNA methylation in prokaryotes
- Maintenance mechanism of 5mCpG methylation
- CpG sequence demethylation reactions in animal cells
- Base excision repair of mismatched base pairs
- Formation of mismatched base pairs and restoration of G:C base pairs through base excision repair
- Passive and active demethylation in early human development
Keywords: CpG sequence, 5-methylcytosine, DNA methyltransferase, S-adenosyl methionine, SAM, CpG island, restriction-modification system (R-M system), maintenance methyltransferase, DNMT1, PAF15, ubiquitination, demethylation, mismatched base pair, base excision repair, TET enzyme, DNA glycosylase (TDG), early development, imprinting genes, epigenetics
2. Characteristics of 5mCpG methylation in animal cell genomes
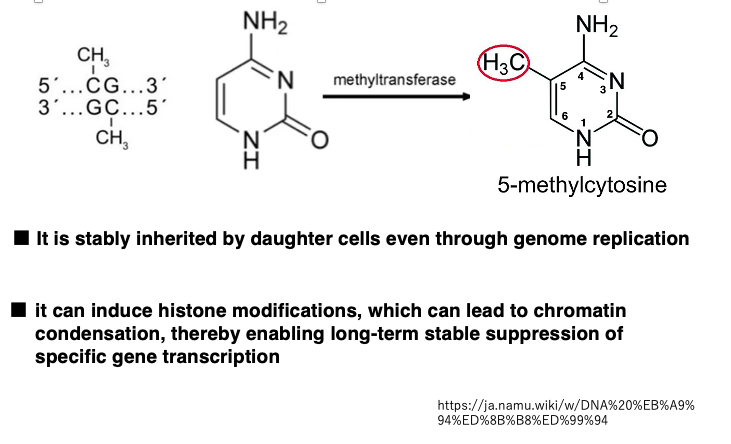
In animal cell genomes, majority of base modifications observed are methylation of the 5th carbon of cytosine within CpG sequences. In 5-methylcytosine, the hydrogen at the 5th carbon position of the cytosine base is replaced with a methyl group. This reaction is carried out by DNA methyltransferase.
In double-stranded DNA, CpG sequences are palindromic. When a CpG sequence on one strand is methylated, the corresponding CpG on the complementary strand is also typically methylated.
Methylated CpG sequences are inherited in DNA strands after genome replication. Additionally, methylated CpG sequences can induce histone modifications, leading to chromatin condensation, which can stably suppress the transcription of specific genes over long periods.
3. The methyl group donor S-adenosyl methionine
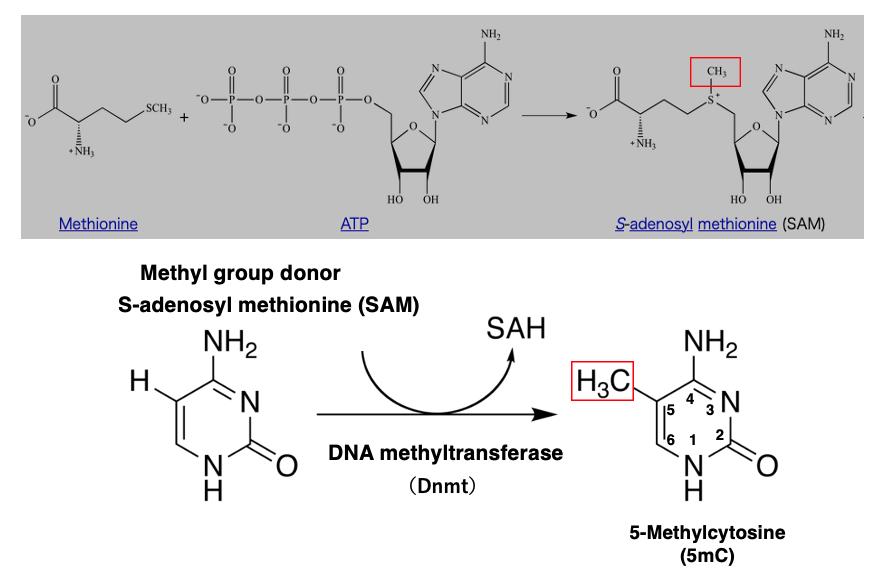
The enzyme that catalyzes the methylation of cytosine is DNA methyltransferase, abbreviated as DNMT.
The methyl group donor used for the transfer is S-adenosyl methionine. S-adenosyl methionine is a complex of the amino acid methionine and ATP, as shown in this figure.
4. Frequency of cytosine methylation in the human genome
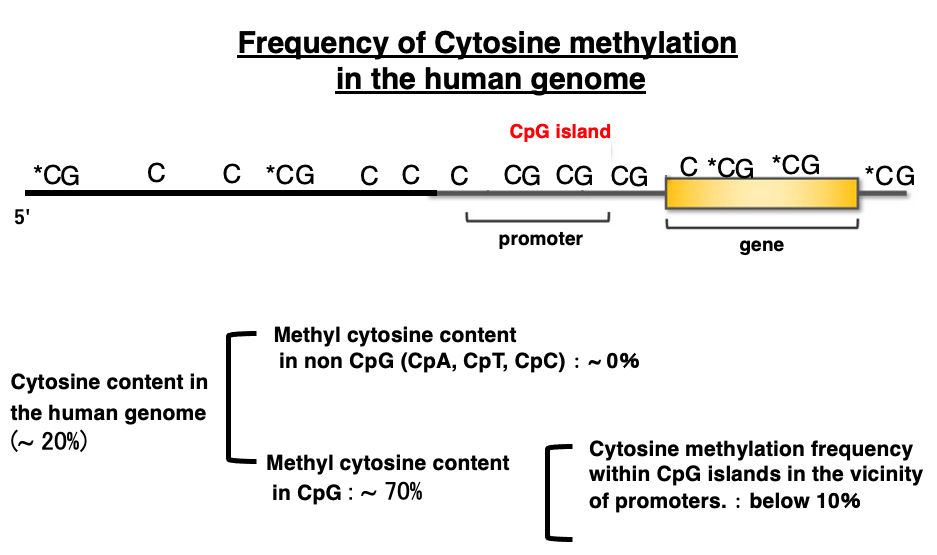
The cytosine content in the human genome is approximately 20%. The methylation of cytosine in CpA, CpT, and CpC sequences is almost zero. On the other hand, the methylation of cytosine in CpG sequences is about 70%.
However, the methylation rate varies greatly depending on where the CpG sequence is located within the genome. In the region near the gene promoter, spanning 500 bp to 3,000 bp, there are more CpG sequences than in other areas, so this region is called a CpG island. Limited to these CpG islands, the frequency of cytosine methylation is low, only about 10%.
5. Comparison of CpG island methylation frequencies based on different genomic positions
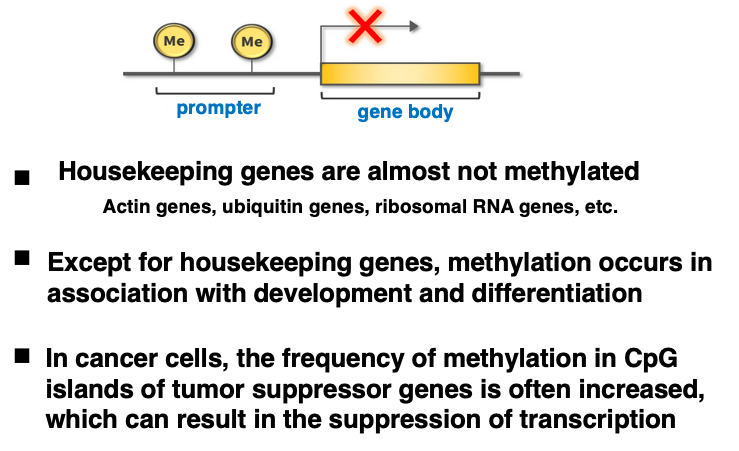
In CpG islands, the frequency of cytosine methylation is generally low, but variations are still observed.
Genes involved in maintaining basic cellular functions are called housekeeping genes. Examples include actin genes, ubiquitin genes, and ribosomal RNA genes. In these housekeeping genes, which are always highly transcribed, the methylation rate of their CpG islands is almost zero.
On the other hand, non-housekeeping genes undergo methylation during development and differentiation. Additionally, in cancer cells, the frequency of methylation in CpG islands of tumor suppressor genes is often high, resulting in suppressed transcription of the genes.
6. CpG suppression in the genome

It has been observed that the occurrence rate of CpG sequences in the genome is significantly lower than the expected rate calculated from the proportion of cytosine in the genome.
The human genome consists of approximately 3 billion base pairs. Since the occurrence rates of cytosine and guanine are both 20%, the estimated number of CpG sequences in the genome is 120 million. However, the actual number of CpG sequences is only 32.28 million, which is merely 1/4 of the expected number based on probability calculations.
This suggests that there has been an evolutionary pressure in the human genome to actively avoid CpG sequences. One possible explanation for this evolutionary pressure is that 5-methylcytosine (5mC) is prone to demethylation within cells, which can lead to its conversion to thymine. If this thymine is not removed before genome replication, it can cause mutations.
7. DNA methylation patterns in land plants and others
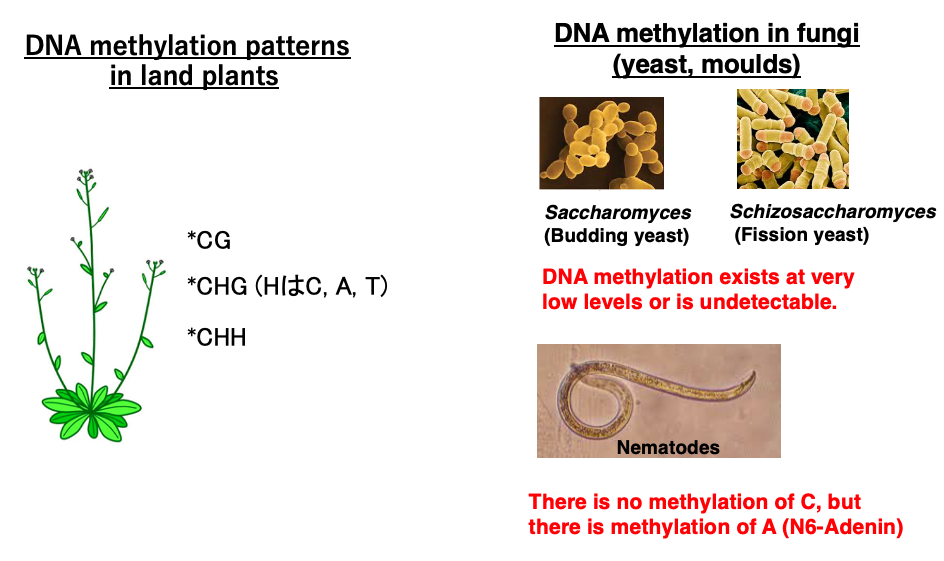
Let’s look at base modifications in eukaryotes other than humans. In plants, in addition to CpG sequences, cytosines within CHG and CHH sequences can be modified to 5mC, similar to animals.
On the other hand, in fungi such as yeast and mold, base modifications are so low that they are barely detectable. Also, in nematodes, there is no methylation modification of cytosine, but adenine undergoes methylation.
8. Transcriptional repression by methylated C in eukaryotes (1)
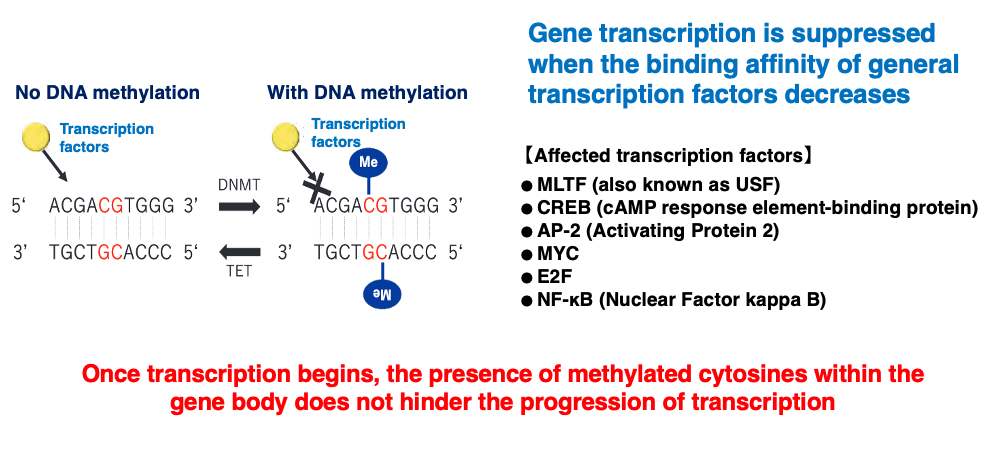
I will now explain the effects of methylated C on transcription. When cytosine in the promoter sequence is methylated, it reduces the binding affinity of transcription factors to the promoter, as shown here. This results in the suppression of transcription frequency.
On the other hand, once transcription has started, the presence of methylated C within the gene body does not hinder the progression of transcription.
9. Transcriptional repression by methylated C in eukaryotes (2)
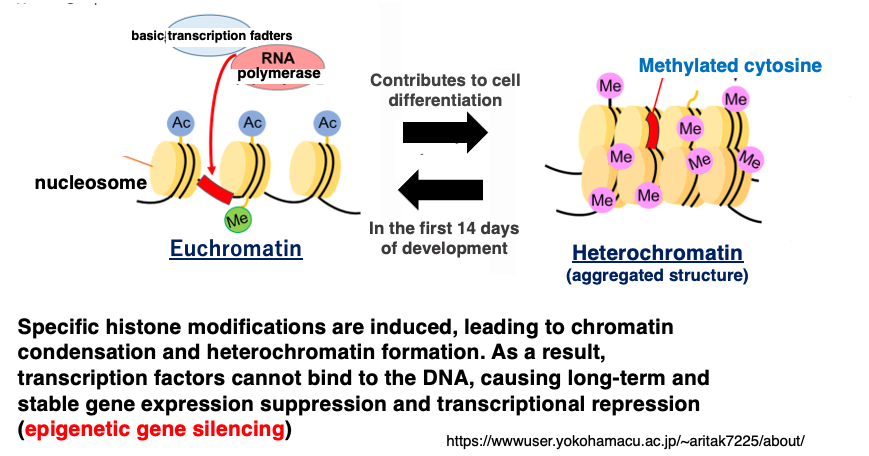
Furthermore, methylation of cytosine induces specific histone modifications. These histone modifications promote chromatin condensation, leading to heterochromatinization, which prevents transcription factors from binding to DNA, resulting in long-term and stable gene expression suppression and transcriptional repression.
This stable and robust suppression of gene expression, formed by the coupling of DNA modification and histone modification, is called epigenetic repression. The term ‘epigenetic’ refers to the phenomenon where genes behave as if they have undergone mutations, even though no actual changes have occurred in the base sequence.
10. Types of DNA methylation in prokaryotes
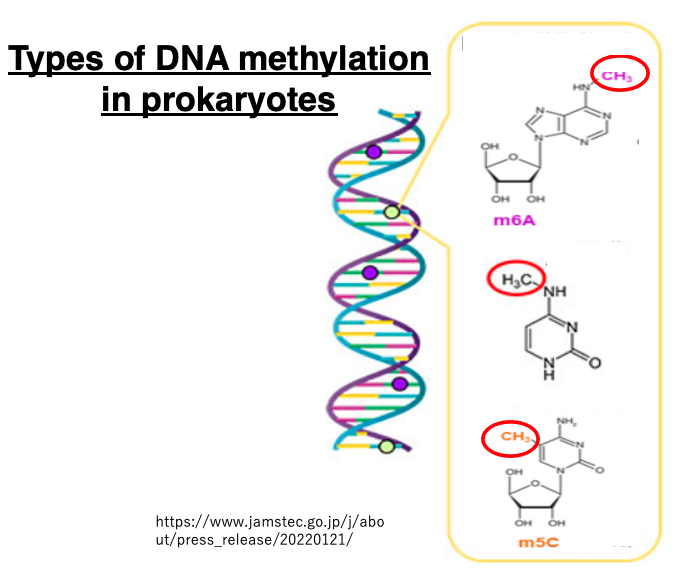
Modified bases can also be found in prokaryotic genomes. These modifications include methylation of adenine and methylation of cytosine.
11. Classification of DNA methyltransferases in bacteria
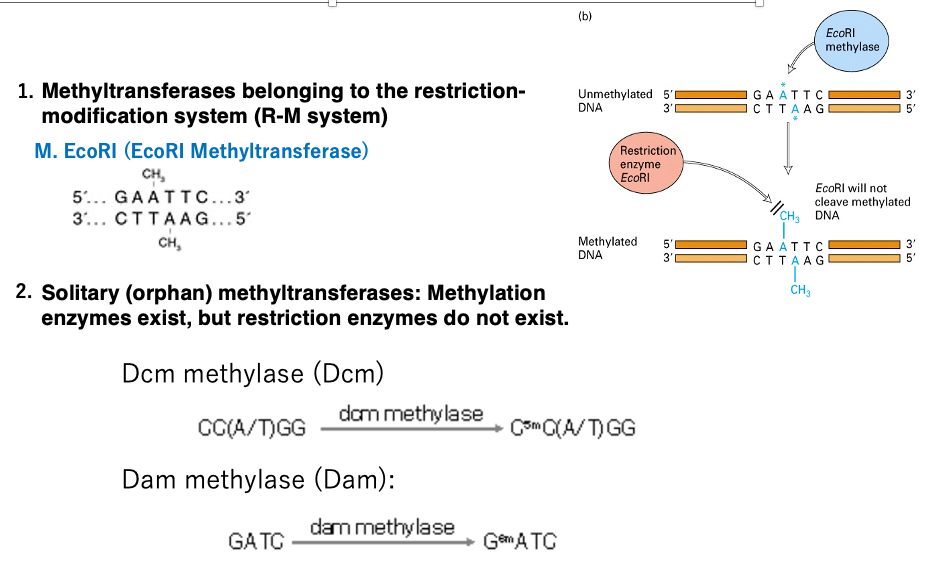
Restriction-Modification (R-M) System: Bacteria possess restriction enzymes within their cells to prevent the invasion and proliferation of parasitic factors such as phages. To protect their own genome from being cut by these restriction enzymes, bacteria also have DNA methylation enzymes that methylate cytosine or adenine within target sequences.
For example, EcoRI methyltransferase methylates adenine in the 5′-GAATTC-3′ sequence, preventing the restriction enzyme EcoRI from cutting this sequence within the bacterial genome.
Orphan Methyltransferases: The second type of methylation enzymes are not part of the restriction-modification system and are called standalone or orphan methyltransferases.Well-known examples include Dcm methyltransferase and Dam methyltransferase. These enzymes introduce methyl modifications to cytosine or adenine within specific sequences, but there are no corresponding restriction enzymes that cut these target sequences.
12. Functions of DNA methylation in prokaryotes
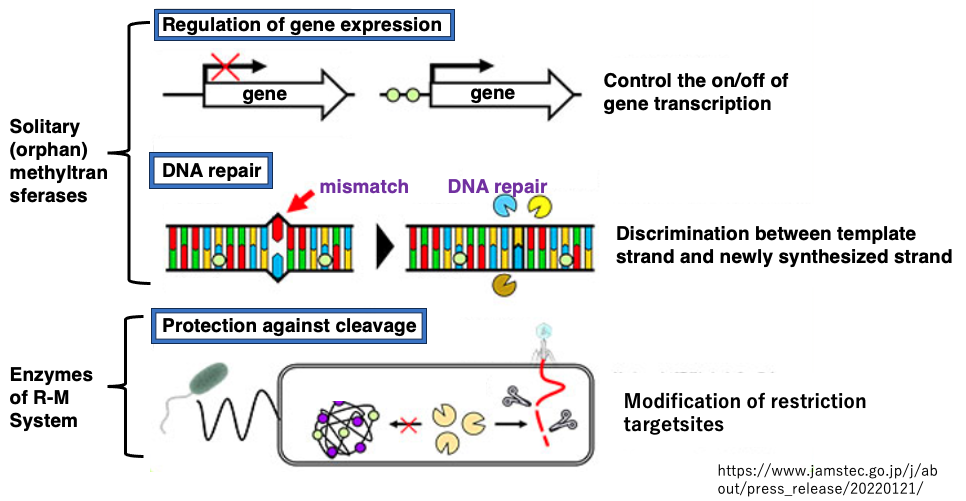
The role of orphan (independent) DNA methylation includes serving as a signal to distinguish between old and new strands in repairing mismatched bases.
13. Mechanisms of maintenance of 5mC within CpG sequences (Transmission to Daughter Cells)
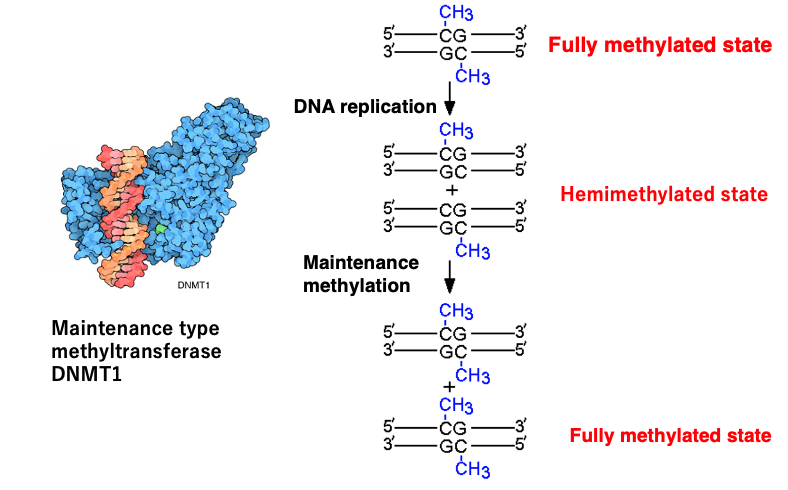
I will explain the mechanism by which methylated cytosines within CpG sequences are maintained after DNA replication.
Before DNA replication begins, both strands of the CpG sequence are methylated, and this state is called the fully methylated state. When DNA strand replication occurs, the methylation of CpG on the old strand remains intact, but there is no methylation on the CpG of the newly synthesized strand. This state is called hemimethylation.
There is a DNA methyltransferase that methylates unmethylated CpG sites on the newly synthesized strand, which in mammals is named DNMT1. DNMT1 is an enzyme that introduces methylation to CpG methylation sites that existed before DNA replication, so it is called a maintenance methylase.
In addition to such maintenance methylases, there are also methylases that introduce methylation to cytosines that have not been methylated before, and these are called de novo methyltransferases (de novo: newly). The mechanism of introducing methylation to new sites on DNA is not included in this lecture.
14. Mechanism of full methylation of *CpG/CpG sites during late S phase
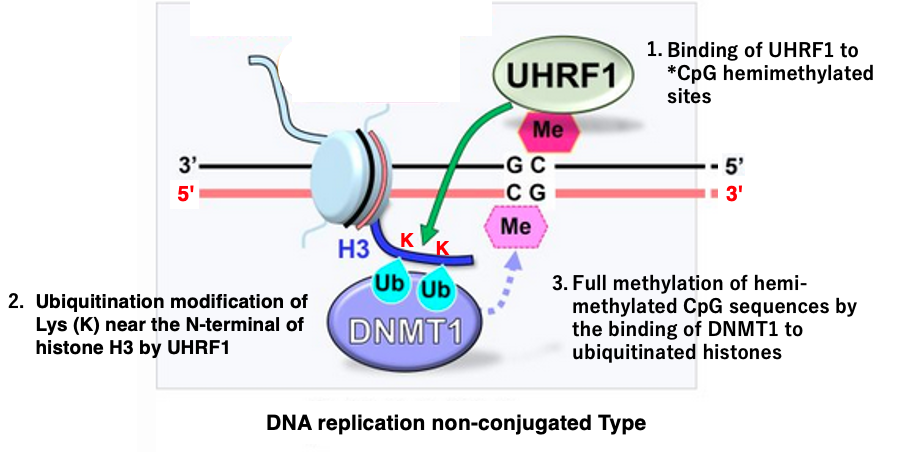
I will explain how the maintenance methyltransferase DNMT1 fully methylates hemimethylated sites. There is a slight difference in the mechanism between early S phase and late S phase of DNA synthesis.
First, let’s discuss the non-replication-coupled full methylation that occurs in late S phase, which is not simultaneous with DNA replication. It would be simple and easy to understand if DNMT1 methyltransferase recognized hemimethylated sites and methylated DNA, but this is not the case.
UHRF1 is a protein that binds to hemimethylated CpG sites. When UHRF1 binds to a hemimethylated CpG site, it ubiquitinates lysine (K) residues near the N-terminus of nearby H3 histones. The ubiquitination of the H3 histone N-terminus recruits DNMT1 methyltransferase, which then methylates the unmethylated CpG.
The ubiquitination of histone H3 by UHRF1 plays a crucial role in facilitating the maintenance of DNA methylation patterns during cell division.
15. Mechanism of full methylation of *CpG/CpG sites during early S phase
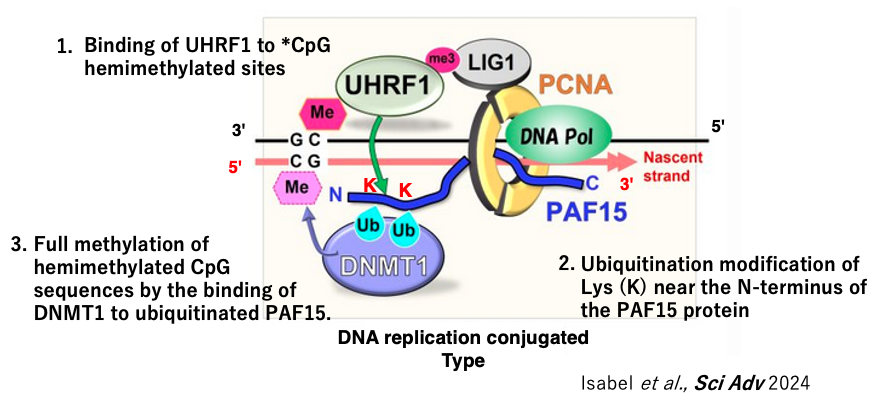
Next, I will explain the methylation of newly synthesized DNA strands coupled with DNA replication during the early S phase.
In this case, UHRF1 attached to the hemimethylated site ubiquitinates the N-terminal Lys of the PAF15 protein, which is part of the DNA replication complex, rather than the H3 histone. This recruits DNMT1 to the PAF15 protein, which then methylates the unmodified CpG. This is the mechanism of replication-coupled CpG methylation maintenance observed in the early S phase.
16. Demethylation reactions of *CpG sequences in animal cells
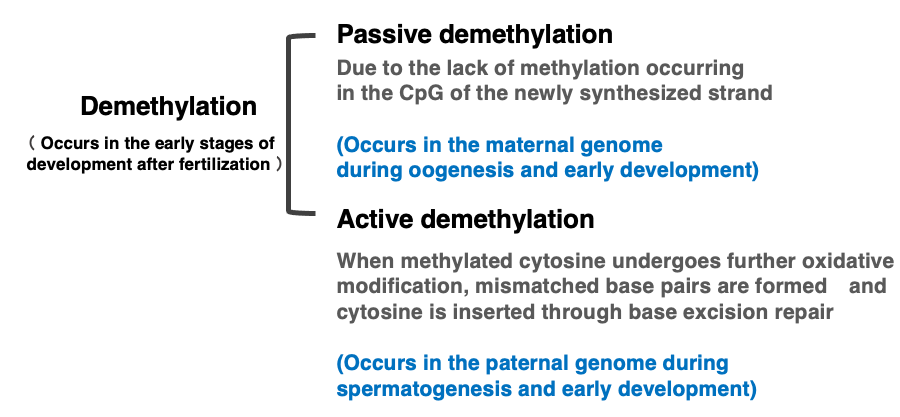
So far, I have explained the function and maintenance mechanism of methylated CpG. Now, I will also explain the mechanism of demethylation of methylated CpG. There are two mechanisms for demethylation of methylated CpG: passive demethylation and active demethylation.
Passive demethylation occurs when the DNMT1 enzyme does not function, resulting in no methylation modification of CpG sequences in the newly synthesized strand. When the DNMT1 enzyme does not work, the number of unmethylated CpG sites increases with each DNA replication. This occurs for the maternal genome in early development.
On the other hand, active demethylation is achieved when 5mC undergoes further oxidative modification, actively forming mismatched base pairs, which are then removed by base excision repair, and cytosine is subsequently inserted. In animal cells, this active demethylation of cytosine occurs during spermatogenesis or in the paternal genome during early development.
17. CpG sequence methylation in white blood cells and sperm
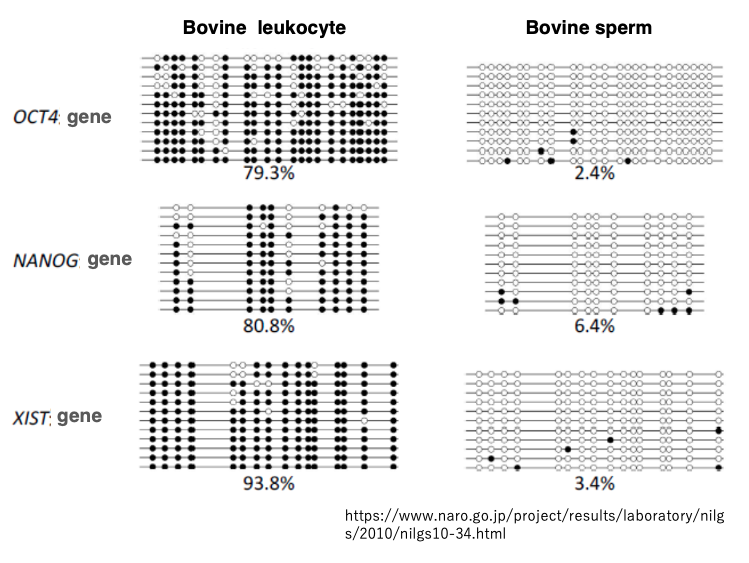
This data shows the analysis of CpG sequence methylation for the OCT4, NANOG, and XIST genes in white blood cells. While sperm is a reproductive cell.
Black circles indicate methylated CpG sites, while white circles represent unmethylated CpG sites.
In white blood cells, we can see that many of the CpG sites are methylated. On the other hand, in sperm, most CpG sites are unmethylated. This indicates that in cattle, extensive demethylation of methylated CpG sites occurs during the process of spermatogenesis.
18. Base Excision Repair of mismatch pase pairs
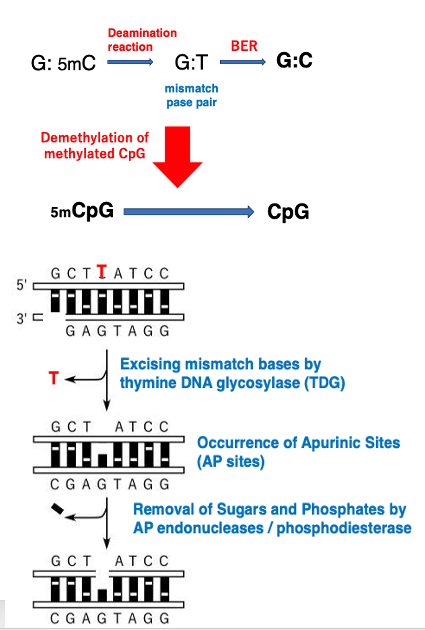
Active CpG demethylation occurs through base excision repair of mismatched base pairs. Here’s an explanation of this process: 5mC (5-methylcytosine) is prone to deamination reaction in cells, which convert it to thymine. As a result, the G:5mC base pair becomes a G:T mismatch.
This mismatched base pair is then processed by thymine DNA glycosylase (TDG), which excises the thymine base, creating an apyrimidinic site (AP site).
After AP endonucleases and phosphodiesterases remove the phosphate group and sugar, DNA polymerase and DNA ligase insert deoxycytidine monophosphate (dCMP). This process effectively demethylates the methylated CpG to an unmethylated CpG.
This mechanism demonstrates how the cell actively removes methyl groups from DNA through a series of enzymatic reactions, ultimately restoring the original cytosine base in the DNA sequence.
19. Formation of mismatched base pairs by TET enzymes and reformation of G:C base pairs through base excision repair
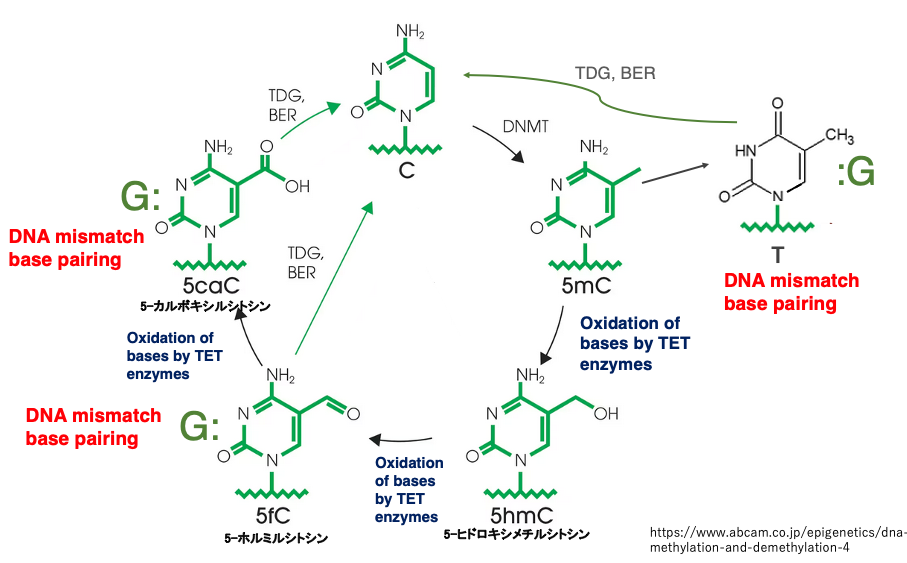
This figure summarizes various methylation modifications of cytosine bases and the formation of mismatched base pairs.
Cytosine is converted to 5mC by the maintenance methyltransferase DNMT1. At this point, the base pairing between G and 5mC is not considered a mismatch, so 5mC is not excised.
When the demethylation system is activated, 5mC is converted to 5-hydroxymethylcytosine (5hmC) and further to 5-formylcytosine (5fC) by TET enzymes that are oxidative enzymes. The pairing of G with 5fC is recognized as a mismatched base pair, so 5fC is excised by thymine DNA glycosylase (TDG) .
Subsequently, dCMP is inserted through base excision repair, resulting in a G:C base pair. If 5fC is not excised, it is further oxidized by TET enzymes to 5-carboxylcytosine (5caC). This is recognized as a mismatched pairing, and through base excision repair, an C is inserted.
In this way, 5mC is oxidized by TET enzymes to create mismatched base pairs, which are then removed by the base excision repair system, resulting in the demethylation of methylated C to cytosine.
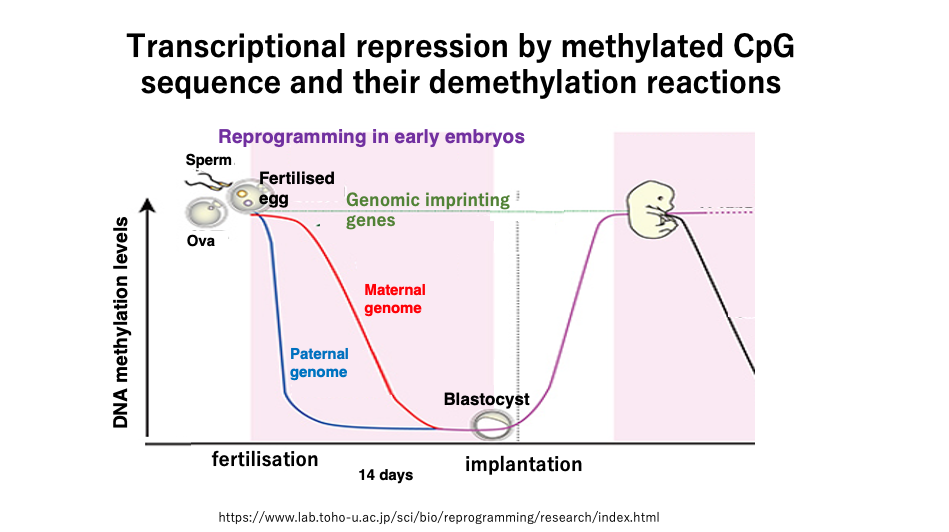
20. Passive demethylation and active demethylation in the early stages of human development
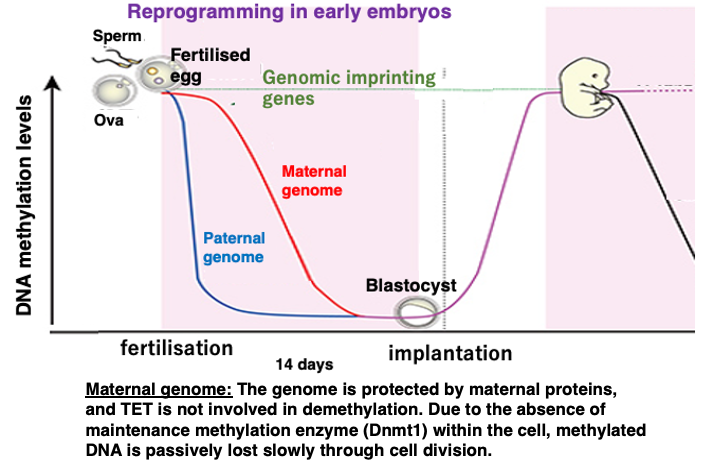
This figure shows the changes in DNA methylation frequency during early human development.
Unlike in cattle, demethylation has not yet occurred during germ cell formation in humans. Extensive demethylation occurs during the first 14 days of development after fertilization.
The maternal genome is protected by maternal proteins, so TET enzymes do not act on it. Also, since there is no DNMT1 enzyme inside the fertilized egg, a slow passive decrease in methylated cytosine frequency occurs through cell division.
On the other hand, in the sperm-derived genome, methylated cytosine is oxidized by TET enzymes, forming mismatched base pairs, which are then removed, resulting in a rapid decrease in the frequency of methylated cytosine.
Even during this early development, there are 100-200 genes that do not undergo demethylation of methylated cytosine, and these are called imprinted genes.
コメント