This lecture can be viewed as a video on YouTube site: Life Scinence Lectures for you
1. Contents of this lecture
In this lecture, I will explain the factors involved in the formation of the three-dimensional structure of proteins, as well as the chaperone molecules involved in protein folding.
1. Primary structure of proteins: alpha helix, beta sheet
2. Primary structure of protein: α-helix, β-sheet, Anfinsen’s dogma
3. α-helix: right-handed helix, one rotation in 3.6 amino acids
4. β-sheet: antiparallel β-sheet (antiparallel), parallel β-sheet (parallel)
5. Tertiary structure of protein: construction of conformation by properties of amino acid side chains
6. Forces acting between side chains: hydrogen bond, ionic bond, hydrophobic interaction, disulfide bond
7. Turn structure: beta-turn, random coil, naturally modified region
8. Quaternary structure of protein: complex of subunits
9. Folding by chaperon help Heat shock and Hsp proteins: Hsp70, Hsp60
10. Trigger factor for folding of nascent proteins 11. Folding by chaperonin complex: GroEL/GroES
Key Words
Αhelix, β-sheet, Anfinsen’s dogma, Chaperon, Chaperonin, Natural denatured region, Protein tertiary structure, GroEL, GroES, Hsp70, Trigger Factor
2. Primary structure of a Protein:Order of Amino acid sequence
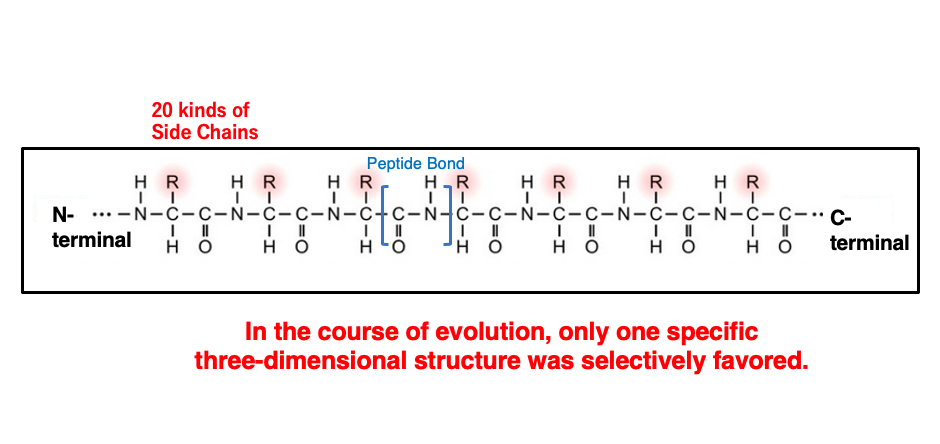
The sequence of amino acids is referred to as the primary structure of a protein. A peptide is a polymer where each amino acid is connected by a peptide bond, and peptides have an N-terminus and a C-terminus. The R-group of each amino acid is called the side chain. For the amino acids used by life on Earth, these side chains are limited to 20 types.
While it may seem that a single peptide chain can take on an infinite number of stable three-dimensional structures, the peptide chain actually designed to adopts a specific single structure. The amino acid sequence of a protein has evolved through natural selection so that only one particular three-dimensional structure has an overwhelmingly much lower free energy compared to others.
As will be discussed later, having two similarly stable structures are harmful to the organism.
3.Prion disease:it is caused by two different three-dimensional structures
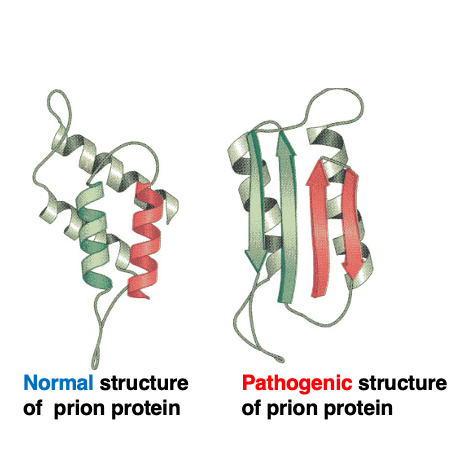
There is an exceptional case where a single peptide can take an equally stable conformation, which is the prion protein. This is the three-dimensional structure of the normal, active prion protein.
On the other hand, this is the three-dimensional structure of the pathogenic, inactive protein. Although these two structures are different, it is known that these two structures are equally stable. Pathogenic protein mainly accumulates and forms aggregates in nerve cells, known as amyloids. In humans, the accumulation of abnormally structured prion proteins leads to Creutzfeldt-Jakob disease, while in sheep it causes a disease called scrapie, and in cattle, it causes bovine spongiform encephalopathy, commonly known as mad cow disease.
In cases where there are two stable conformations, one is normal while the other lacks proper activity, and the accumulation of the abnormal protein causes harm. Even correctly structured proteins, due to factors like heat, can undergo denaturation and change to an abnormal conformation. Such denatured proteins can also cause the formation of aggregates, so cells have systems to promptly degrade them, such as proteasome system.
4. Anfinsen’s Dogma : Princeple of protein folding
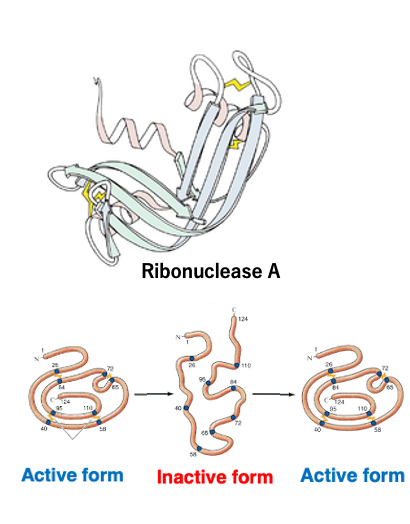
Anfinsen, a British researcher, found that by heating ribonuclease A to disrupt its three-dimensional structure and inactivate it, and then slowly removing the heat, the original active structure was restored.
Based on such experimental results, Anfinsen proposed the hypothesis that a protein’s native structure is uniquely determined by only its amino acid sequence, and that this structure represents the most thermodynamically stable state. This is known as Anfinsen’s dogma.
The dogma is fundamentally considered correct – a protein’s three-dimensional structure is specified by its amino acid sequence, and no other information is essentially required.
5. Peptide bond in the protein backbone : it is arranged in the same plane
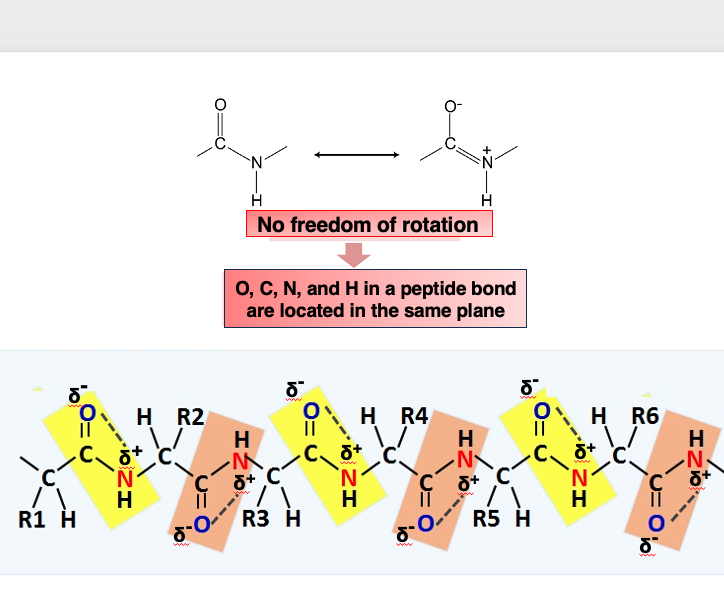
The peptide bond in the protein backbone consists of a carbonyl group with a partial negative charge (δ-) at the oxygen atom, and the nitrogen atom of the amide group having a partial positive charge (δ+), which attract each other. As a result, the C, O, N, and H atoms involved in a peptide bond cannot freely rotate, and they are arranged in the same plane.
Therefore, when observing the protein backbone, the peptide bonds are arranged in the same plane, while the atoms forming the side chains can freely rotate.
Regarding the secondary structure of proteins, there are structures called alpha helices and beta sheets. Let me first explain the alpha helix structure.
6. A type of protein secondary structure :α-helix
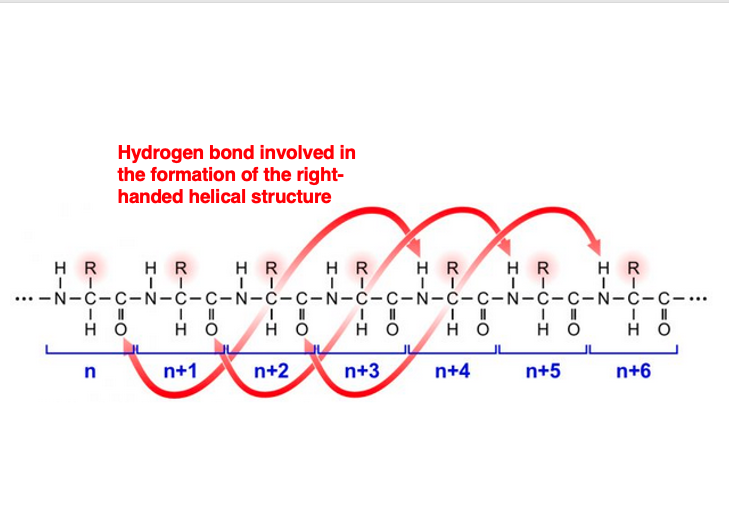
This is the carbonyl group of the Nth amino acid residue that forms the main chain. This carbonyl group’s oxygen forms a hydrogen bond with the amino group’s hydrogen of the number (n+4) amino acid residue.
The carbonyl group of the number (n+1) residue forms a hydrogen bond with the amino group of the number (n+5) residue, and so on. As a result, every amino acid residue participates in hydrogen bonding.
This leads to the formation of a right-handed helical structure. This is called the alpha-helix structure.
7. Alpha helix structure : pitch length of 0.54 nm
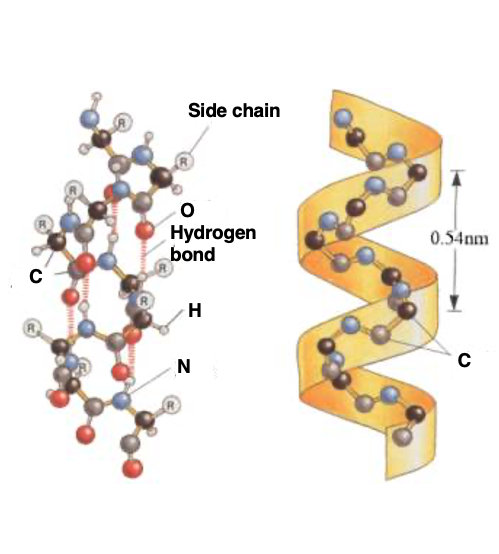
To explain the same alpha helix with another diagram, it looks like this. Hydrogen bonds are regularly formed between the carbonyl and immune groups, and the side chains, which are the R groups, protrude outward. This right-handed helical structure with a pitch length of 0.54 nm, from here to here, is the alpha helix structure.
8. α-helix : right-handed helical coil structure with 3.6 amino acid residues per turn
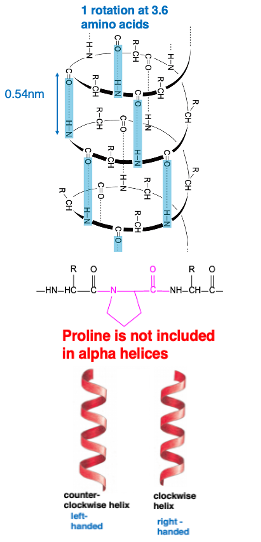
This is a schematic diagram of the alpha helix. Hydrogen bonds are regularly formed between the carbonyl groups and amino groups. A hydrogen bond is formed between the number N carbonyl group, and the number (n+4) immuno-group. As a result, a right-handed helical structure appears, making one turn every 3.6 amino acid residues.
Regarding right-handed and left-handed, this figure shows a right-handed helix, and this figure shows a left-handed helix. Imagine a nail screw-like object, if you rotate it clockwise, it moves forward that is right-handed. Therefore, this is a right-handed helix. In the case of the alpha helical structure of proteins, it is a right-handed structure, and left-handed structures are generally not formed.
It is known that the amino acid proline is not included in the alpha helical structure. Proline is a very special amino acid, lacking an amino group in it.
In peptide bonds containing proline residues, the proline residue forms a cyclic structure, so this part cannot freely bend. Therefore, at the positions where there is a proline residue in the polypeptide chain, the alpha helix is not properly formed. Conversely, this means that proline residues are not included in alpha helices.
9. β pleated sheet (β sheet)
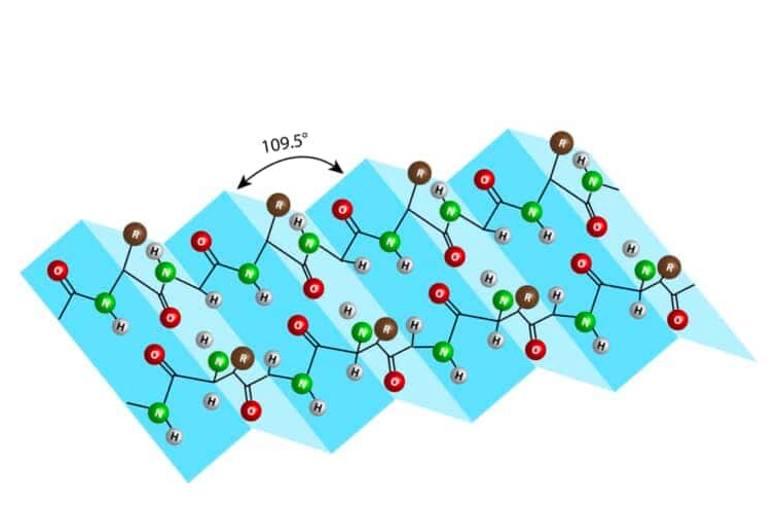
The second type of secondary structure in proteins is called the β-pleated sheet or β-sheet.
It is formed by hydrogen bonds between the carbonyl and immune groups of two extended polypeptide strands. The term “pleated” refers to the pleats or folds in a skirt. Each fold is arranged at an angle of 109.5 degrees, creating a pleated or folded structure. This is what is known as the β-pleated sheet or β-sheet secondary structure.
10. Antiparallel and Parallel β sheets
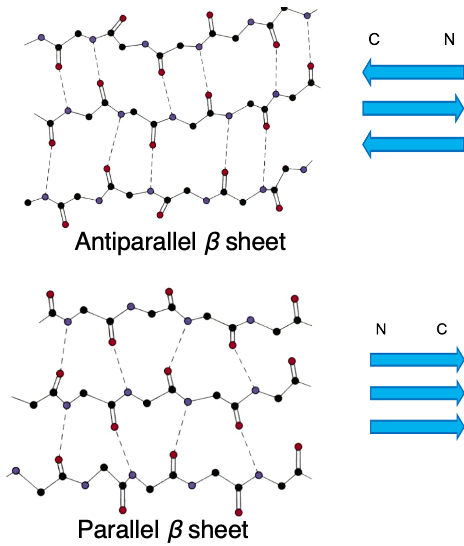
There are two types of β-sheets, anti-parallel β-sheets and parallel beta sheets. Proteins have an N-terminus and a C-terminus, making them directional polymer molecules.
Anti-parallel β- sheets are formed when hydrogen bonds are formed between main chains with opposite directionality, while parallel β- sheets are formed when hydrogen bonds are formed between main chains with the same directionality.
11. Anti-parallel parallel β-sheet
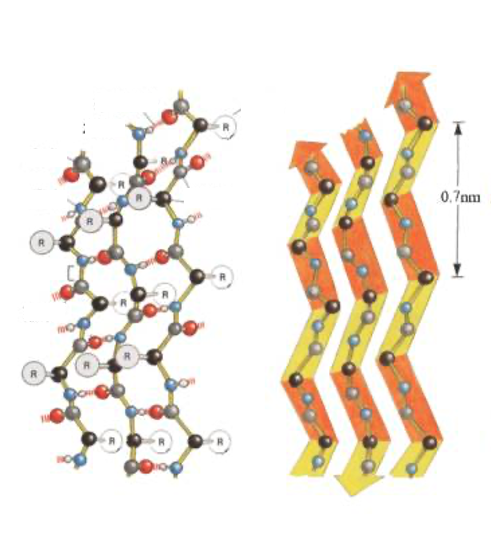
This figure also shows the Anti-parallel parallel β-sheet. Hydrogen bonds are formed between peptide chains with different orientations, resulting in this flat folded structure.
12. Tertiary structure of protein
Next, I will explain the tertiary structure of proteins. This is the three-dimensional structure formed by the combination of alpha helices and beta sheets explained earlier, and is called the tertiary structure. While the alpha helix and beta sheet structures are formed by hydrogen bonds between the carbonyl and immuno groups of the peptide chain, the tertiary structure of proteins is strongly influenced by the side chains of each amino acid.
The properties of the side chains are the major determinants of how the protein folds. In other words, the folding of the peptide occurs such that amino acids with hydrophobic side chains are positioned on the inside, while amino acid residues with hydrophilic side chains are positioned on the outside.
Therefore, even though amino acids may be far apart in the primary sequence, they can become proximal in the tertiary structure. So, herefore, if an antibody is obtained using a native protein as antigen, the antibody may not be able to detect the denatured protein in Western blotting, because the epitope should be recognized by the antibody may have been destroyed.
13. Factors determining tertiary structure
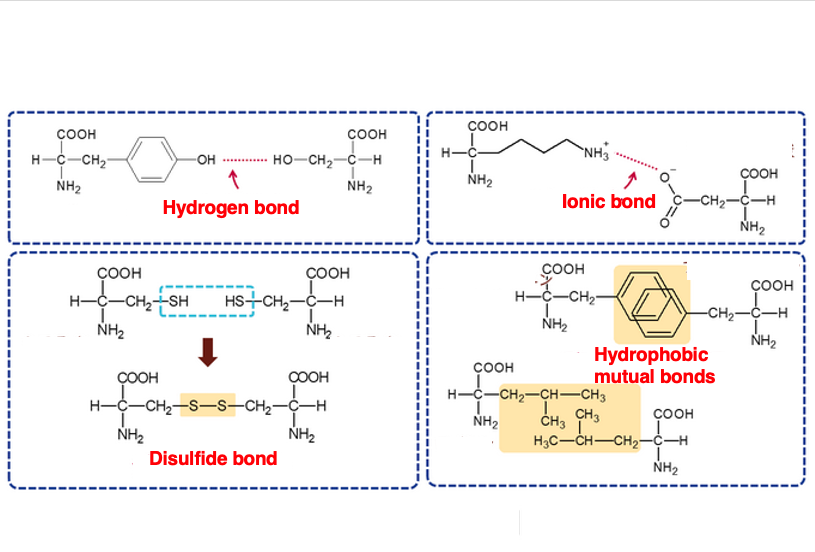
This summarizes the interactions between side chains that determine the three-dimensional structure.
The first is hydrogen bonding. The second is ionic bonding, and the third is hydrophobic bonding. The fourth is the S-S bond between SH groups. Among these four interactions, this is the only covalent bond. Such four kinds of interactions between the side chains are the main factors that determine the three-dimensional structure.
The cytoplasm is in a reduced state, so S-S bonds cannot form inside the cell. However, when a protein is secreted outside the cell, it is exposed to an oxidizing environment, so S-S bonds between cysteine and cysteine rapidly form, giving the protein an active three-dimensional structure.
14. βturn : turn structure consisting of 4 amino acids
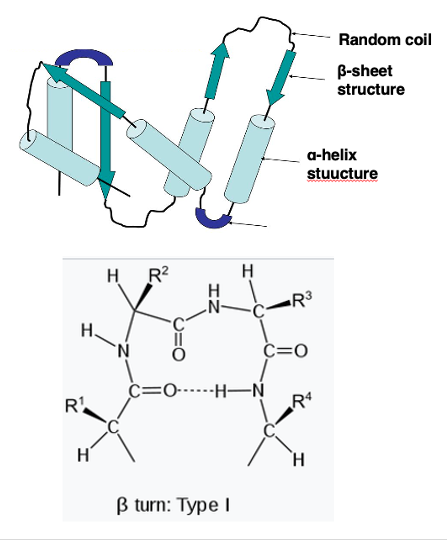
Let me further explain other parts of the three-dimensional structure. The regions connecting α-helices to other α-helices, or α-helices to β-sheets, are called turn structures.
The most popular type of turn structure is a β-turn. A β-turn consists of four amino acid residues. Here, four amino acid residues are shown. A hydrogen bond forms between the carbonyl group of the first residue and the immuno group of the fourth residue, resulting in the β-turn structure.
It is known that amino acids like proline and glycine are often found in such turn structures.
15. Intrinsically Disordered Regions within protein
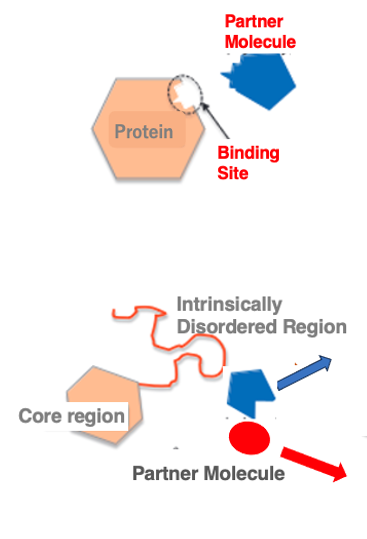
Within three-dimensional structure of protein, regions that do not have a well-defined structure are called random or intrinsically disordered regions. It has long been well-known that when two proteins interact, their contact surfaces have complementary shapes that fit together like a lock and key.
However, recent studies have revealed that even interacting proteins may not have pre-formed structures resembling a lock and key. Analysis of protein structures shows that in addition to well-structured core regions, there are regions lacking a defined three-dimensional structure. These regions are called intrinsically disordered regions. Such disordered regions only form a rigid structure upon binding to a partner molecule.
The partner is not limited to a single specific molecule; within the cellular environment, these regions can form complexes with different partner molecules. The disordered regions can flexibly change their structure depending on the binding partner. The proportion of such disordered regions in proteins is estimated to be 16-22% in eukaryotes and 3-7% in prokaryotes.
16. Quaternary structure of protein
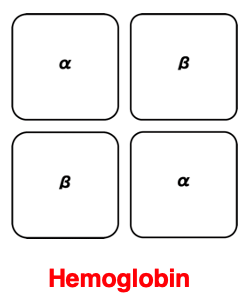
Let me briefly touch on the quaternary structure of proteins. When 3D structured polypeptide chains associate into multiple complexes, allowing them to perform specific functions more efficiently, this complex of polypeptide chains is called the quaternary structure of the protein.
One of the most well-known examples of a quaternary structure is hemoglobin. In the case of hemoglobin, it consists of two alpha globin proteins and two beta globin proteins, forming an α2β2 hetero-tetramer. This allows it to efficiently bind and release oxygen. This concludes the explanation on protein 3D structure, then move to a related topic.
17. Chaperone molecules that assist protein folding
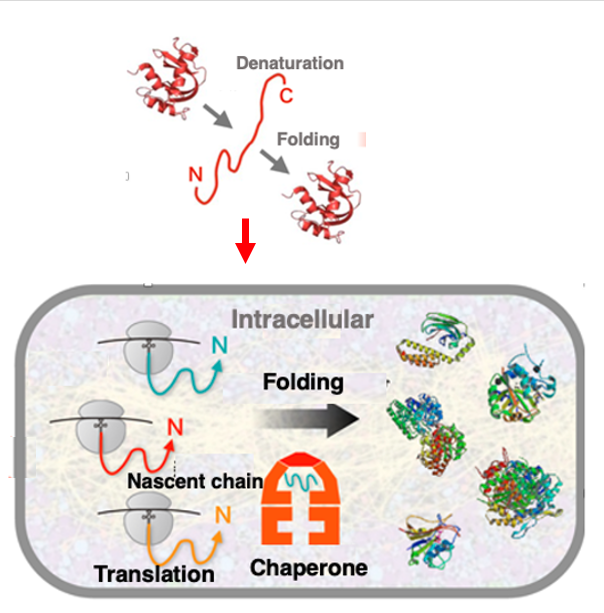
Anfinsen demonstrated that RNase A could regain its active three-dimensional structure in vitro after being denatured by heat, without any external assistance factors. This led to Anfinsen’s dogma that proteins can automatically adopt the lowest energy conformation determined by their amino acid sequence, without external help.
However, the situation is slightly different inside the cell cytoplasm, where protein concentrations range from 300 to 350 mg/mL, creating a highly crowded environment. In such conditions, newly synthesized proteins must rapidly fold into their correct structures; otherwise, they risk aggregating due to unintended interactions with surrounding proteins.
To rapidly attain the lowest energy conformation dictated by their amino acid sequence within the crowded cellular environment, the assistance of proteins called chaperones is essential. As soon as the N-terminus of a nascent polypeptide emerges from the ribosome’s large subunit, chaperone-assisted folding begins. This allows newly synthesized proteins to fold correctly without aggregating, despite the high concentration of various proteins inside the cell.
Initially studied chaperone molecules include the heat shock proteins (HSPs), designated by their molecular weights in Daltons, such as HSP60 and HSP70. These proteins were first identified as a group strongly expressed in bacteria like E. coli when exposed to high temperatures around 42°C. Further research revealed that HSPs help refold proteins denatured by heat shock and prevent their aggregation.
Today, chaperones are generally defined as proteins that assist in proper folding of peptides and refolding of denatured, aggregated proteins, often coupled with ATP hydrolysis‘s.
18. Heat shock protein : Early chaperone molecules studied in the research history
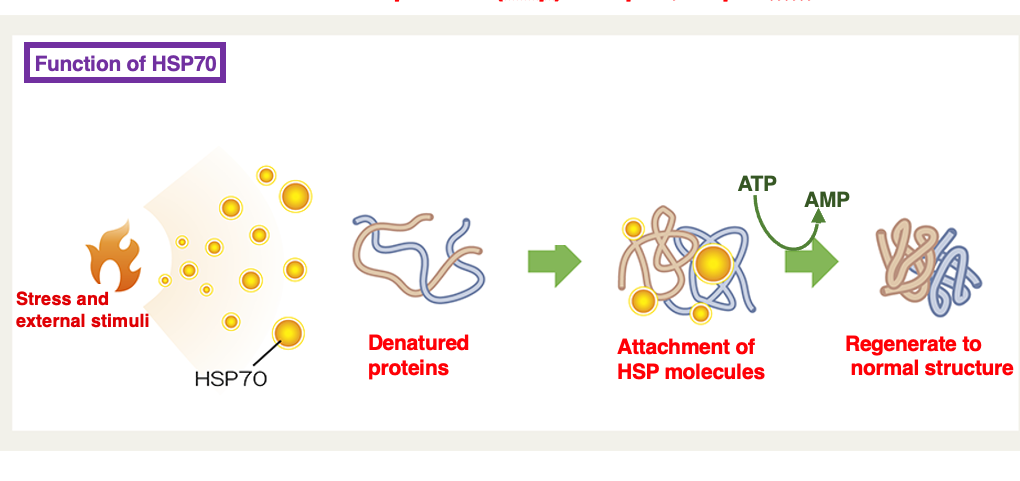
Heat shock proteins are belonging to chaperone proteins that have been studied very early. The proteins are designated by their molecular weights in Daltons, such as HSP60 and HSP70. These proteins were first identified as a group strongly expressed in bacteria like E. coli when exposed to high temperatures around 42°C.
Further research revealed that HSPs help refold proteins denatured by heat shock and prevent their aggregation. Today, chaperones are generally defined as proteins that assist in proper folding of peptides and refolding of denatured, aggregated proteins, often coupled with ATP hydrolysis‘s.
19. Trigger Factor : A type of chaperone
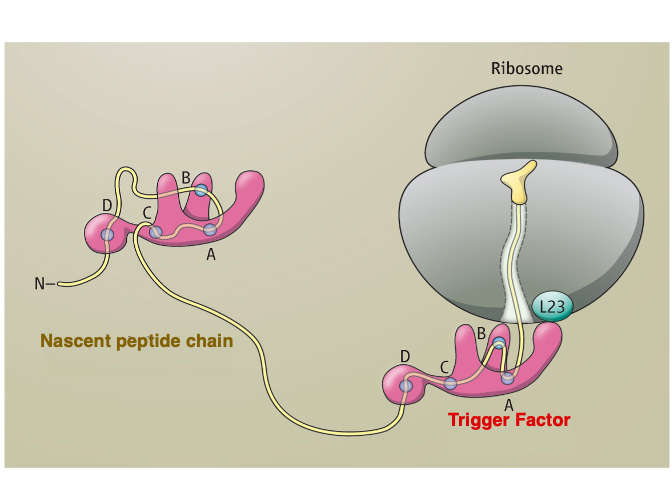
20. GroEL/GroES Chaperonin
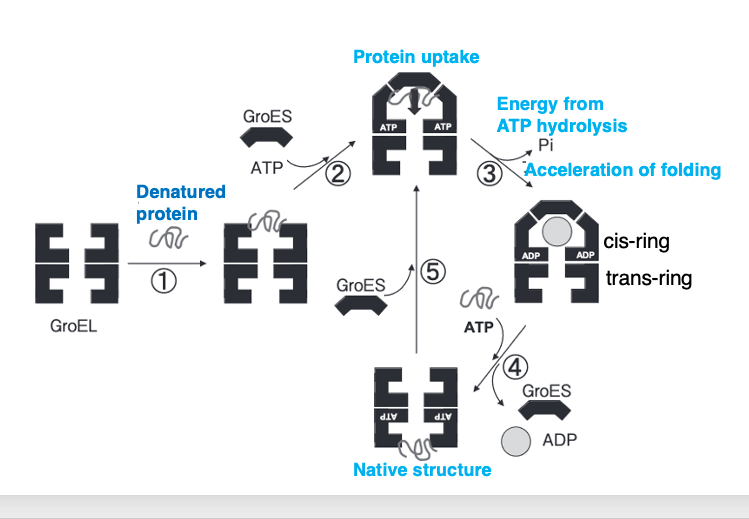
In E. coli, a complex of two chaperone proteins, GroEL and GroES, is used to assist in the folding of newly synthesized proteins and the refolding of denatured proteins.
The GroEL-GroES complex is often specifically referred to as a chaperonin among chaperone molecules. Denatured proteins are taken up into GroEL. When a denatured protein enters GroEL, GroES acts as a lid on the cavity formed by GroEL.
Within this enclosed space, using ATP, the denatured protein or the newly synthesized protein that has not yet formed the correct three-dimensional structure is properly folded. Once the protein is correctly folded, the GroES cap dissociates, and the protein is released from GroEL.
21. Degradation system for unrepairable proteins : proteasomal degradation
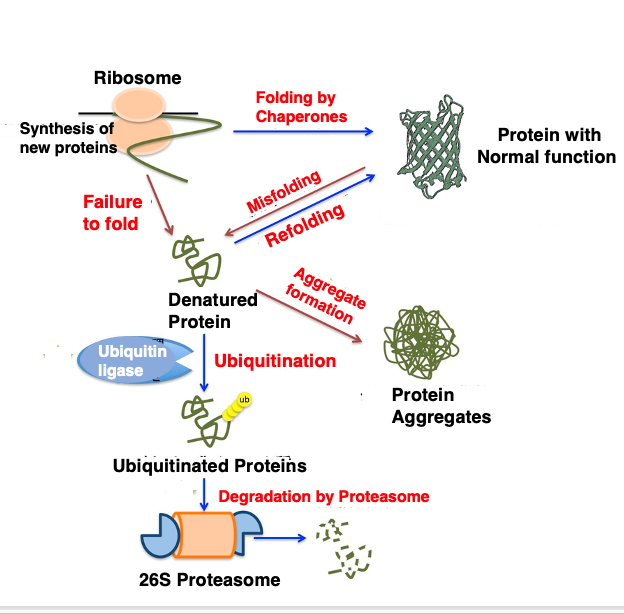
As I have explained, within cells, efforts are made to properly fold newly synthesized proteins using chaperones, or to refold misfolded proteins using chaperones.
However, if denatured proteins cannot be properly refolded, these improperly structured proteins can aggregate and form insoluble clumps. These aggregates can have harmful effects on the cell. For example, accumulation of protein aggregates within nerve cells can cause diseases like Alzheimer’s and Huntington’s disease.
To prevent this, cells tag proteins that cannot be properly refolded with a small peptide called ubiquitin, and then, send these proteins to a large protein degradation machinery called the proteasome, where they are degraded.
This concludes the explanation about protein structures. See you again soon.
コメント