Contents of this lecture can be viewed on YouTube site : Life Science Lectures for You
https://youtu.be/iSUmjqK21dw
1. This lecture includes the following topics:
- The three-dimensional structures of two prion proteins
2. Kuru disease in Papua New Guinea
3. Transmission of disease by misfolded prion proteins
4. Mechanism of prion disease onset
5. Cause of the spread of mad cow disease
6. Prion-like proteins found in yeast
Key words: Prion, Creutzfeldt-Jakob disease (CJD), kuru, amyloid plaques, bovine spongiform encephalopathy (BSE), mad cow disease, meat and bone meal
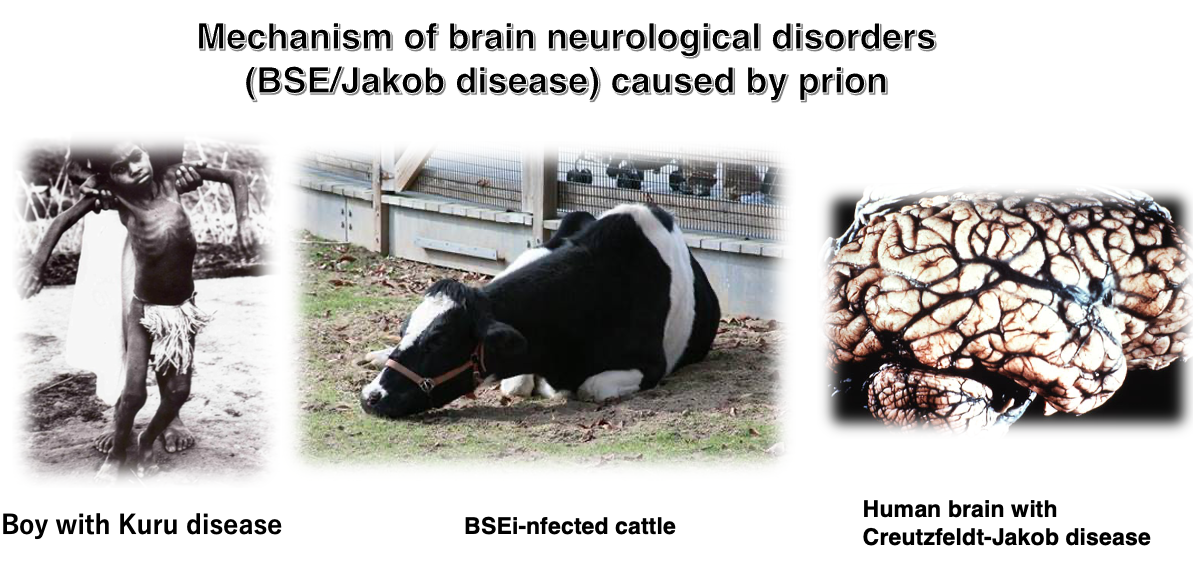
2. Mechanism of brain neurological disorders (BSE/Jakob disease) caused by prion

I will explain about prion disease. It is a disease that occurs due to the existence of two stable three-dimensional protein structures, can be transmitted between individuals of the same species, and in some cases, can even cross species barriers to transmit the disease. It is a prion disease.
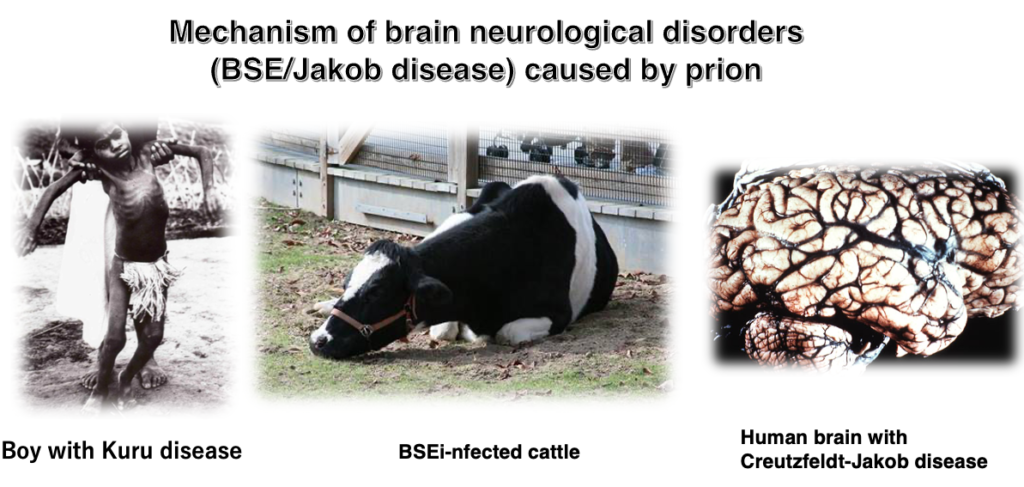
3. Prion diseases in various mammals
This table shows the prion diseases known in mammals. In humans, prion diseases are generally referred to as Creutzfeldt-Jakob disease (CJD). Additionally, a disease called Kuru, which was endemic in certain regions of New Guinea, is now known to have been a form of Creutzfeldt-Jakob disease. In other animals, prion diseases are known by different names: in sheep, it’s called scrapie; in cattle, it’s bovine spongiform encephalopathy (BSE), commonly known as mad cow disease, which caused worldwide panic. Prion diseases also occur in deer, mink, cats, and other animals. Although the symptoms of these diseases vary slightly depending on the species, it is now clear that they are all caused by the same protein called prion protein.
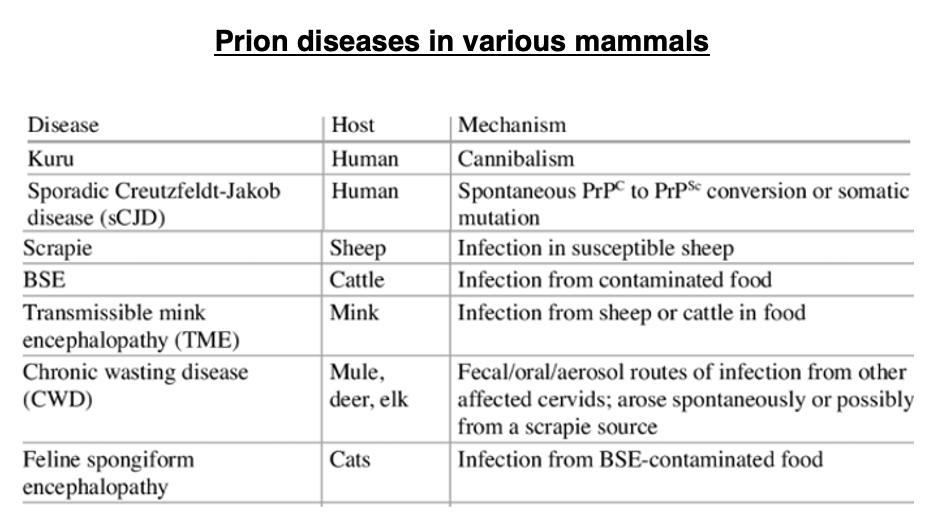
4. Principles of protein tertiary structure: The existence of a single three-dimensional structure that is significantly lower in energy than all others
I would like to first review some general concepts about protein folding into three-dimensional structures. This is commonly referred to as Anfinsen’s dogma, which posits that once the amino acid sequence of a protein is determined, it will fold into a three-dimensional structure with the lowest free energy through interactions between side chains, given ideal environmental conditions. It is believed that the amino acid sequence has been evolutionarily selected to form a structure with significantly lower energy compared to other possible structures.
Furthermore, it is believed that the amino acid sequence has been evolutionarily selected in such a way that the resulting three-dimensional structure possesses significantly lower energy compared to other possible structures.
5. The three-dimensional structure of normal and abnormal prions
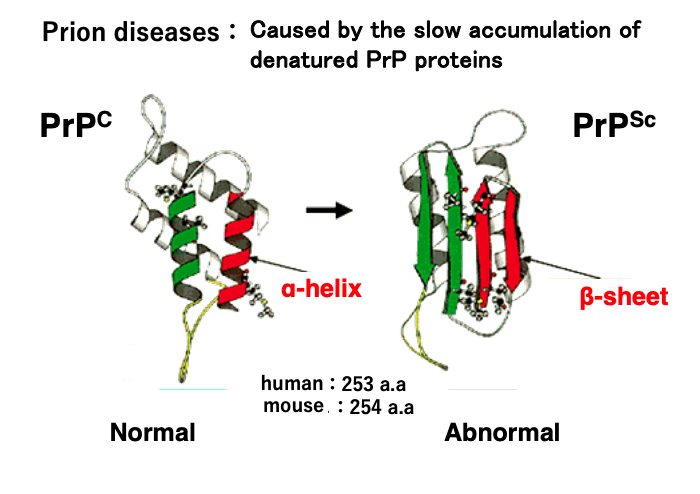
In prion diseases, the abnormally structured prion protein is denoted as PrPSc. It is now known that the disease is caused by the slow accumulation of this prion protein in cells without being degraded.
The ‘Sc’ designation originates from ‘scrapie,’ a prion disease in sheep characterized by the animal’s behavior of scraping against fences. The figure on the left shows the normal structure of the prion protein, labeled as PrPC. The ‘C’ stands for ‘cellular prion protein’.
The abnormally structured prion protein is formed when, for some reason, two α-helices (shown in green and red in the left figure) of the normal prion protein are converted into β-sheets, resulting in a structurally abnormal protein. The human prion protein is a relatively small protein consisting of 253 amino acids with a molecular weight of 27,000 daltons.
6. Formation of amyloid plaques due to accumulation of abnormal prions
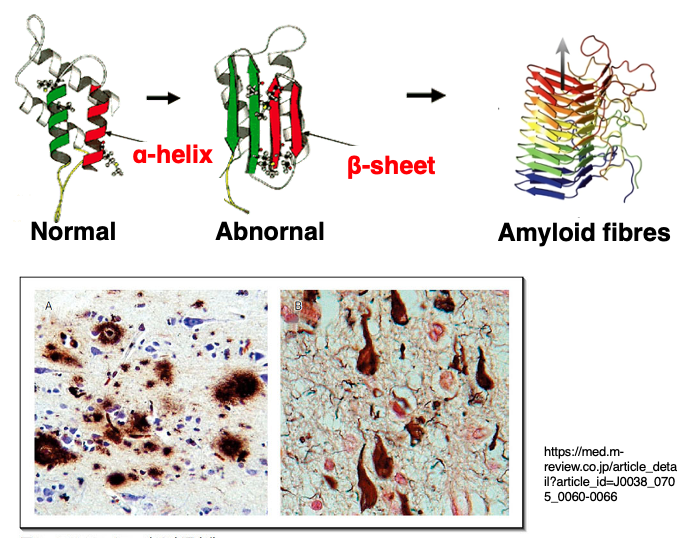
Prion proteins that have undergone structural abnormalities aggregate to form amyloid fibrils or amyloid plaques. The β-sheets of abnormal prions interact with each other, leading to aggregation and formation of protein clumps called amyloid plaques or amyloid fibrils.
Since prion proteins are normally highly expressed in neuronal cells, the accumulation of abnormal proteins occurs prominently in brain and nervous system tissues. Once the accumulation of abnormal proteins begins, the process cannot be interrupted and ultimately leads to the death of the individual.
7. Endemic disease of Papua New Guinea: Kuru
I will explain the process of how the mechanism of prion disease development was elucidated.
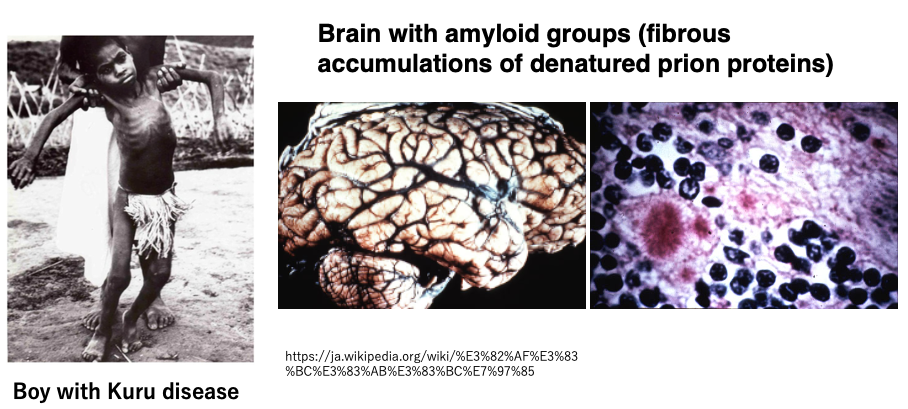
Although it is now recognized as a prion disease, it was discovered in the 1950s as a neurological endemic disease (a disease that occurs in specific regions or populations) affecting the Fore people of Papua New Guinea. This endemic disease was named Kuru.
The boy in the photo has Kuru and cannot walk properly. At that time, it was recorded that there were 2,584 patients among the Fore people, of which 70% were adult women, very few (10%) were adult men, and 20% were children. Among adults, there was a characteristic high ratio of women and few men.
Today, we know that the cause of Kuru was due to the Fore people’s custom of eating the flesh of humans who had developed Kuru, primarily by women, for religious reasons.
8. The substance that causes Kuru disease: A protein without nucleic acids?
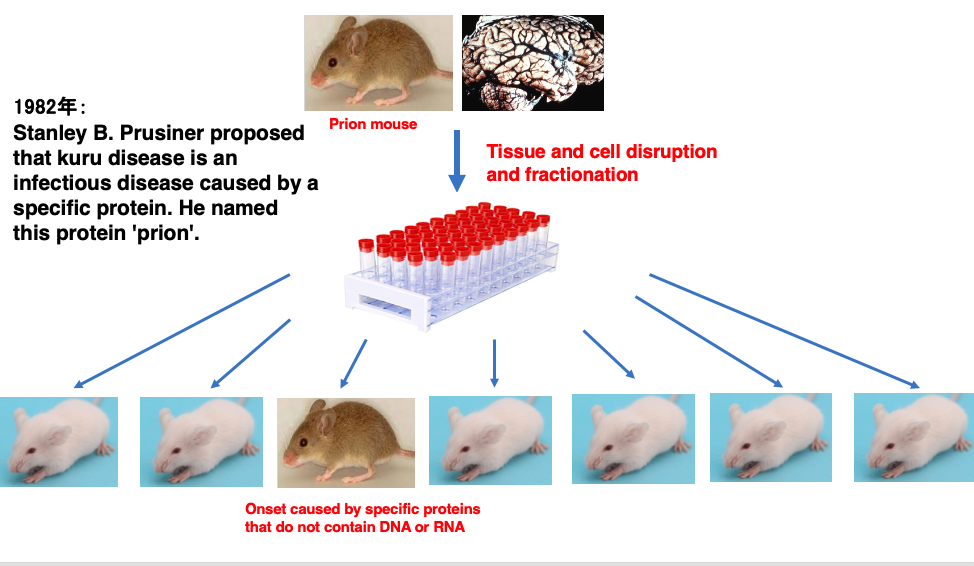
Research was initiated to investigate the mechanism of Kuru disease onset. As cases of mice exhibiting symptoms similar to Kuru disease were discovered, experiments were primarily conducted using mice.
Brain tissue was extracted from mice with Kuru disease, and the cells were crushed and separated into components such as proteins, nucleic acids, and lipids. These components were then administered to healthy mice. As a result, it was experimentally determined that after a long incubation period, mice developed Kuru disease, albeit at a low frequency.
Through such experiments, in 1982, Stanley Prusiner proposed that Kuru disease is an infectious disease caused by a specific protein. He named this specific protein ‘prion’. This experiment captures the key points about the research into Kuru disease, Prusiner’s groundbreaking proposal about prions in 1982.
9. The prion protein is encoded on chromosome 2
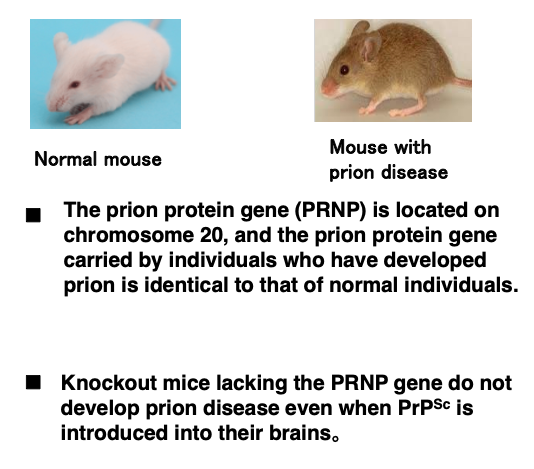
Subsequent research revealed that the gene encoding the prion protein, called the PRNP gene, is located on chromosome 2 in humans. However, in mice, it was found that the sequence of the prion gene in individuals that developed prion disease was identical to that of normal individuals, indicating that DNA sequence mutations are not the cause of prion disease.
Furthermore, it was demonstrated that knockout mice lacking the prion gene do not develop prion disease even when abnormal prion protein is directly introduced into their brains or administered in their feed.
10. A protein without nucleic acids as the cause of infection?
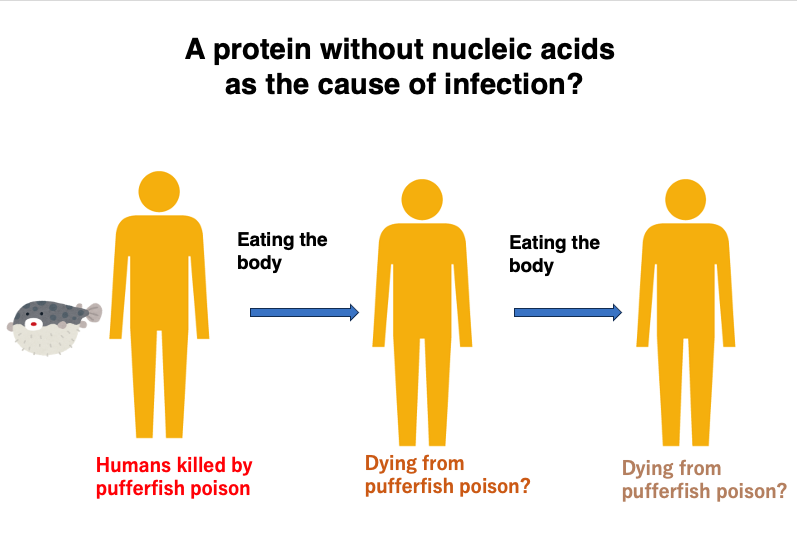
Researchers were greatly perplexed by such experimental results.
The fact that a pure protein without genetic material like DNA or RNA could be the cause of prion disease, act as a source of infection, and spread the infection to other individuals was difficult to explain with the molecular biology knowledge of the time.
For example, if someone were to die from eating a poisonous pufferfish, it’s unlikely that another person would die from eating part of the deceased person’s body. Even if they did die, it’s inconceivable that a chain reaction would occur where someone else would die from eating the person who died from eating the first victim. This is simply because protein toxins would be diluted as they pass from human to human.
If the cause were a replicating factor with genes, like bacterial or viral infections, it could spread infection indefinitely from individual to individual without dilution, as it would replicate within newly infected individuals. So, researchers couldn’t conceive of a mechanism by which a pure protein could cause infection continuously over a long period, spreading from individual to individual.
11. The mechanism by which misfolded prion proteins can transmit disease from one individual to another
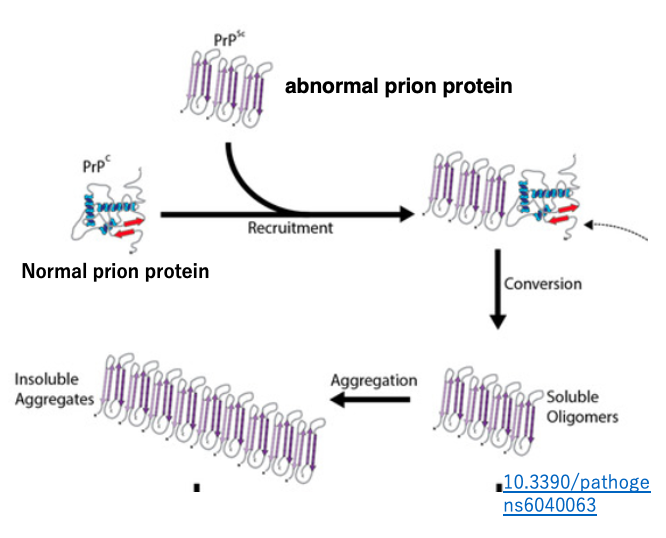
The possible mechanism has been discussed and hypothesized by researchers from various fields. The proposed model suggests that the misfolded prion protein acts as a seed, interacting with existing normal prion proteins and altering their three-dimensional structure to become misfolded.
According to this model, normal prions with a proper structure are continuously supplied from normal genes, so if a single molecule of misfolded prion is introduced into the body, it will gradually increase in number and accumulate within cells that are producing the normal proteins.
12. Indigestibility of abnormal prion proteins
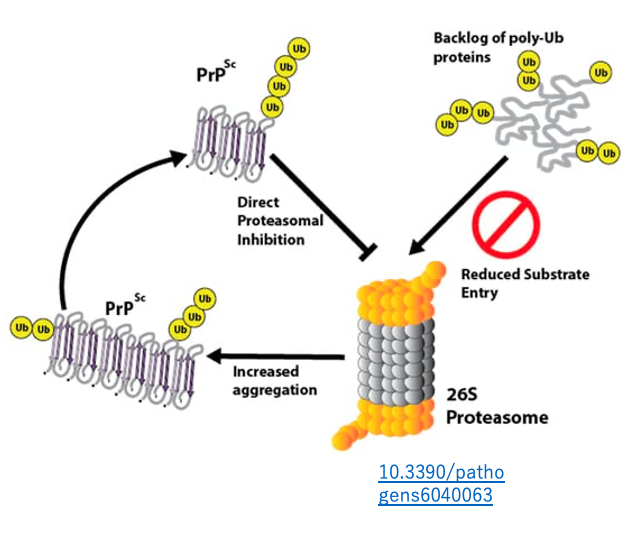
Generally, abnormal proteins are tagged with a short peptide chain called ubiquitin and degraded in the proteasome. However, it has been discovered that abnormal prions bind to the proteasome and inhibit its function. Consequently, when abnormal prions are present, other abnormal proteins also become difficult to degrade in the proteasome. As a result, abnormal proteins accumulate increasingly within cells, eventually leading to symptoms such as difficulty walking.
This is the mechanism of prion disease onset. The incubation period is long, considered to be at least 8 to 10 years. Moreover, the fatality rate after onset is 100%, with death occurring about one year after symptoms appear.
13. Global epidemic of bovine prion disease, mad cow disease (BSE)

Here’s a brief explanation of bovine spongiform encephalopathy (BSE), commonly known as mad cow disease. BSE was first reported in the United Kingdom in 1986. The disease got its official name “bovine spongiform encephalopathy” because the brains of affected cattle had a sponge-like appearance. In English, it’s abbreviated as BSE, standing for Bovine (cattle) Spongiform (sponge-like) Encephalopathy (brain disease).
As shown in this image, cattle infected with BSE display neurological abnormalities and are unable to walk normally. The BSE epidemic began on British farms in the late 1980s and early 1990s, eventually spreading throughout Europe and even reaching North America. In 2001, the first case of BSE was confirmed in Japan. The peak of the epidemic was in 1992, when approximately 37,000 cattle worldwide were found to be infected. Between late April 1996 and the end of 2001, about 5.3 million cattle were culled globally.
14. How BSE spread rapidly worldwide : Use of meat and bone meal from BSE-infected cattle
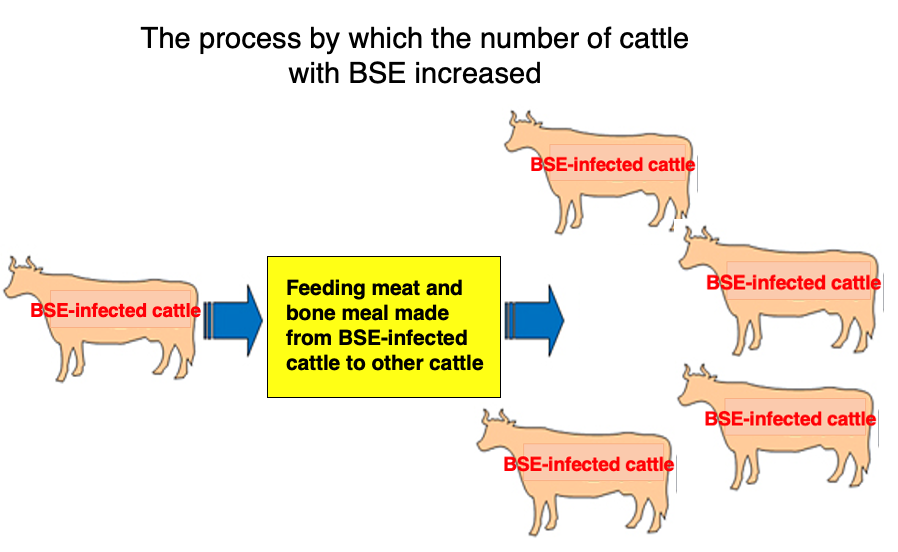
The rapid spread of BSE worldwide can be attributed to the following process: In every country, meat from cattle infected with BSE cannot be used for human consumption. As a result, these infected cows were processed into meat and bone meal, which was then used as a raw material for cattle feed. When healthy cattle consumed meat and bone meal from BSE-infected cows, the abnormal prion proteins were ingested by the healthy cattle. This triggered the accumulation of abnormal prions in previously healthy cows, following the mechanism described earlier.
Complicating matters further, abnormal prion proteins are difficult to digest in the stomach and retain their properties even when heated. These characteristics of prions derived from BSE-infected cattle in meat and bone meal contributed to the spread of the disease. It is believed that when ingested orally, the abnormal prion proteins, without being digested, enter through the lymphoid tissue in the intestines and then invade the brain via nearby nerve fibers.
15. Human transmission of BSE
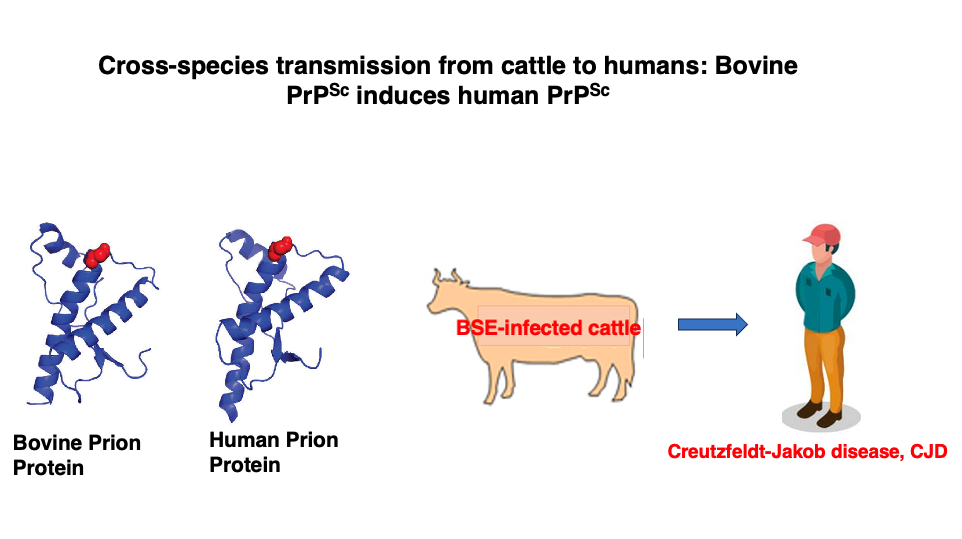
The proliferation of cows with BSE is problematic in itself, but the situation became even more serious when it was discovered that BSE could be transmitted from cattle to humans.
It has been found that consuming meat from BSE-infected cattle, especially meat near the spinal cord and brain where prion protein accumulation is significant, can lead to the ingestion of altered bovine prion proteins into the human body. Additionally, abnormal prion proteins are highly resistant to heat, making it difficult to inactivate them through normal heating processes. To inactivate them, heating at 134°C for 18 minutes is necessary. Three-dimensional structure of the abnormal prions are so stable.
These abnormal prions can then transform normal human prions into their pathogenic form. The discovery of this cross-species transmission from cattle to humans has greatly made worse the issue.
16. The end of prion disease outbreaks and the natural incidence rate

These newspaper articles are from 2001 when the first case of a cow infected with BSE was discovered in Japan. While BSE shocked the world, the disease eventually subsided globally after establishing rules prohibiting the use of infected cattle in meat and bone meal production.
While the BSE epidemic in cattle has been largely controlled, sporadic cases of CJD continue to occur naturally in human populations at a low rate. It affects approximately 1 individual per million per year worldwide, with about 350 cases diagnosed annually in the United States. The disease is invariably fatal, with most patients dying within a year of onset. A healthy person who receives a corneal transplant from a patient with naturally occurring CJD may also develop the disease.
Unfortunately, there is currently no cure for CJD, and it remains an incurable condition. Researchers are hoping to develop treatments such as vaccines that stimulate the production of antibodies that bind only to abnormal prion proteins.
17. Prion-like proteins that also exist in yeast
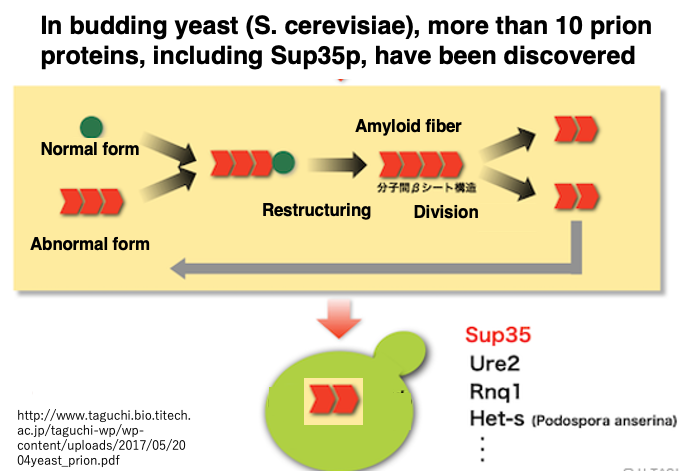
In recent years, prion proteins have come to be understood in a broader sense. They are now widely regarded as proteins that have a catalytic effect, acting on normal proteins to alter their three-dimensional structure and transform them into a different stable conformation. In this broader definition, prions do not necessarily need to share homology with the prion proteins that cause prion diseases in humans and cattle.
Under this expanded definition, the yeast Saccharomyces cerevisiae contains more than 10 types of prion proteins, including SUP35. This means that in yeast, at least 10 types of proteins have been found that can act on normal proteins, convert their structure to the same abnormal form as themselves, and accumulate within cells.
Moreover, in the case of yeast, the accumulation of abnormal proteins does not seem to be necessarily detrimental to the cell. In principle, a protein’s three-dimensional structure is overwhelmingly stable in only one conformation compared to others, which is essential for maintaining an orderly metabolic system in living organisms. However, it has become known that in certain yeast growth environments, the accumulation of such abnormal proteins may actually be advantageous for survival. Similar proteins might also be found in higher organisms.
コメント